
Hazard Analysis and Critical Control Points (HACCP) is a science-based system that identifies, evaluates and controls hazards of significance to assure food safety. Simply put, the focus of Hazard Analysis is that hazards and appropriate control measures are identified and Critical Control Points (CCPs) are further delineated as control measures essential to eliminate or reduce the hazard to an acceptable level. The boundaries that separate acceptability from unacceptability are defined as critical limits.
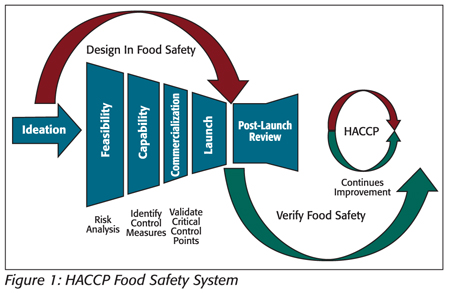
Where manufacturing processes and control measure components have been predefined in product and process design, the application of HACCP can be a reflective process for the facility HACCP team as they evaluate each control measure in determining “What hazard is of significance and is the step specifically designed to eliminate or reduce the likely occurrence of a hazard to an acceptable level?” (see Figure 1).
Aseptic processing and packaging refer to the processing and packaging of a commercially sterile product into sterilized containers followed by hermetically sealing with a sterilized closure to prevent viable microbiological recontamination.
Hazard Analysis and Control Measures
For low-acid (pH > 4.6), shelf-stable, nonrefrigerated beverages, heat-resistant spores of toxigenic anaerobic microorganisms, such as Clostridium botulinum, are a biological hazard of significance that warrant absolute control. Foodborne botulism can be severe, resulting from the ingestion of foods containing the neurotoxin formed during growth of the organism if present and allowed to grow in the product. Thus, the primary strategies in minimizing C. botulinum risk are in having effective control measures for (i) sterilization and maintaining sterility of the processing equipment; (ii) destruction of heat-stable C. botulinum spores in product; (iii) sterilization of the packaging material and (iv) maintaining sterility during filling and packaging.
For HACCP, the components of these essential control measures can be defined as CCPs. For a regulated scheduled process, these control measures can be described as critical factors.
A critical factor is defined by the U.S. Food and Drug Administration (in 21 CFR 113.3) as any property, characteristic, condition, aspect or other parameter, a variation of which may affect the scheduled process and the attainment of commercial sterility.[1] The ‘scheduled process” means the process selected by the processor as adequate under the conditions of manufacture for a given product to achieve commercial sterility.
This article provides some HACCP examples of control measures in which critical components and limits have been predefined in the design of a process to ensure commercial sterility for low-acid beverages.
Equipment Sterilization
Sterilization is a process aimed at the complete destruction of microorganisms and their spores. Before production start-ups, all components of the process equipment downstream from the sterilizer hold tube must be brought to a condition of commercial sterility and maintained during production to ensure commercial sterility. Typically, the equipment components would include an ultra-high temperature (UHT) sterilizer, all holding tanks and lines after the UHT sterilizer and the filler. For each of the components, Hazard Analysis and control measure evaluations by the HACCP team should take into consideration the time and temperature required for the coldest part of the process to meet sterilization parameters and that calibrated monitoring equipment is located appropriately to indicate desired performance. A common industry guideline is using the performance criteria of time and temperature to achieve inactivation of bacteria and bacteria spores (i.e., >121 °C for 30 min.).
The process authority may indicate additional critical factors to the equipment manufacturer for the process. A process authority is a competent person having expert knowledge of aseptic processing and packaging for making determinations of a scheduled process.
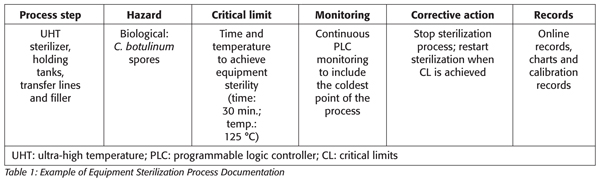 Corrective actions, when the sterilization temperature and time fail to reach the critical limit, are controlled by the equipment through automatic stoppage (Table 1). The sterilization program should be reset and the machine restarted. Checking the temperature monitoring devices prior to start-up will verify that the system is functioning; however, this should not be considered as a validation that is required during the process commercialization.
Corrective actions, when the sterilization temperature and time fail to reach the critical limit, are controlled by the equipment through automatic stoppage (Table 1). The sterilization program should be reset and the machine restarted. Checking the temperature monitoring devices prior to start-up will verify that the system is functioning; however, this should not be considered as a validation that is required during the process commercialization.
Product Sterilization
The process authority, in conjunction with the equipment manufacturer, defines the scheduled process, taking into consideration critical parameters, such as incoming spore load, product formula, pH, rheology, heat penetration, flow rate, residence time and equipment surface contact area.[2] The typical acceptability for the process is often defined by a multiple of 12 for the D value (i.e., the time required at a certain temperature to kill 90 percent of the organism) of C. botulinum, or its equivalent. To compare thermal processes calculated for different temperatures, a standard Fo value is assigned for each product. This Fo value is the time in minutes (at a reference temperature of 250 °F and with a z = 18 °F) to provide the appropriate spore inactivation to achieve commercial sterility. The sterilization parameters are usually both product and process specific.
In a continuous product flow process, the time for which the product must be held at the defined temperature to attain sterility is achieved in the section of the hold tube. The flow rate of each particle of the hold tube is critical. It is essential that the rate of flow for the fastest particle or the shortest particle retention time be accurately determined for each product flow rate, length, dimension and design of the hold section and product type and characteristics. The use of dye or salt injection can be employed to determine minimum residence time. Mathematical models that incorporate the flow rate, product rheology and the dimensions and design of the hold tube are used to calculate the minimum residence time required to achieve product sterility. For situations in which flow characteristics are unknown, experimental design studies may be used to validate the thermal process.
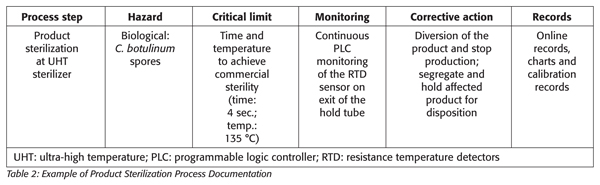 Corrective actions, when the sterilization temperature and time fails to reach the critical limit, automatically divert the product for reprocessing or destruction (Table 2).
Corrective actions, when the sterilization temperature and time fails to reach the critical limit, automatically divert the product for reprocessing or destruction (Table 2).
Packaging Sterilization
The objective of packaging sterilization is the same as for equipment sterilization: the destruction of bacteria and spores on packaging surfaces to ensure commercial sterility for cold filling and packaging of the product. The packaging material, preformed containers and their closures are usually sterilized inside the packaging machine or externally and introduced aseptically into the aseptic zone of the packaging machine. For sterilization inside the packaging machine, it is usually accomplished by heat or through a combination of chemical and physical treatments.[3]
In the example of using hydrogen peroxide for packaging sterilization, most of the validation and performance acceptance levels are conducted by the manufacturer, leaving the final validation to be conducted by the commercialization facility and verified by the HACCP team (e.g., using Bacillus subtilis for modeling temperature and time requirements for equipment and packaging sterilization). The critical factors, defined by the equipment manufacturers, may include sterilant concentration, mode of application, temperature, contact time and packaging contact surface size with acceptance criteria of 4–5 log reduction for spores. Additionally, there may be other regulatory limits such as the minimum concentration of 30% with residual hydrogen peroxide regulated at a maximum level of 0.5 ppm.
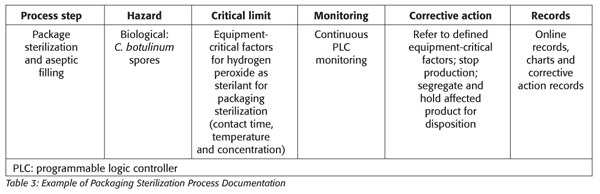 Corrective actions for the packaging and filling operations of these complex systems are often predefined by the equipment manufacturer and the process authority (Table 3).
Corrective actions for the packaging and filling operations of these complex systems are often predefined by the equipment manufacturer and the process authority (Table 3).
In addition to the above equipment and process steps, the HACCP team should conduct Hazard Analysis and evaluate control measures that are essential to maintaining process sterility. The evaluation should include components such as steam barriers, overpressure and associated HEPA filtration systems.
Verification
The facility HACCP team, during the HACCP reviews, verifies the critical limits that control the specific hazards and ensure product safety. The verification process may be performed by an individual who is qualified in the particular field and is not responsible for the routine monitoring of the critical limits. One such example is verification of the reliability of the results through calibrations being performed to acceptable international standards by an independent third party, thus showing that measuring devices are accurate and precise.
Measurement of critical limits in each of the components of the scheduled process can be verified independently, for example, divert checks performed on the UHT sterilizer at the start-up of production to show that product that has undergone insufficient temperature treatment will be diverted away from filling. Another effective method for verifying the effectiveness of product sterilization is through media fill trials, where a microbiologically sensitive medium, such as Linden Grain, is sterilized, aseptically filled into sterile packs and plated for total viable count, yeast and mold. The microbial content of the media-filled packs must show an absence of growth.
Conditions required for the sterilization of filling machines can be verified by placing Bacillus stearothermophilus spore strips of known spore loads in target locations and measuring the log reduction following equipment sterilization.
Alarms on filling machines with a hydrogen peroxide immersion system can be tested to show they sound when the hydrogen peroxide bath level is below a minimum volume. A more quantitative method for verifying the effectiveness of packaging sterilization is by inoculation pack testing, where preformed packaging is inoculated with a known amount of bacterial spores and introduced into the packaging machine where it is sterilized. The packaging then undergoes microbiological testing to determine the log reduction post-sterilization. Results from package integrity testing where filled packs are inspected for leakages or incorrect sealing can also verify that the product has been hermetically sealed at the filler. Record keeping is an important part of the verification process, as it serves as documented evidence that critical limits have been reviewed and verified to be fully functional at maintaining aseptic control.
Summary
Low-acid aseptic beverage systems represent a highly complex sector of the food industry. Historically, these processes represent a successful story, as most of the food safety design requirements are predefined by equipment manufacturers and the process authority in the commercialization process with final verification by the HACCP team. A strong partnership must be maintained among all parties, as there can be little room for error when C. botulinum is the primary hazard.
 Suchart Chaven is a food safety director for PepsiCo AMEA (Asia, Middle East & Africa) in Dubai.
Suchart Chaven is a food safety director for PepsiCo AMEA (Asia, Middle East & Africa) in Dubai.
 Ana Sedarati is a microbiology and sanitation manager at PepsiCo AMEA in Dubai.
Ana Sedarati is a microbiology and sanitation manager at PepsiCo AMEA in Dubai.
2. Code of Hygienic Practice for Aseptically Processed and Packaged Low-Acid Food. CAC/RCP 40-1993.
3. Ansari, M.I.A. and A. K. Datta. 2003. An Overview of Sterilization Methods for Packaging Materials used in Aseptic Packaging Systems. Trans ChemE 81:57–65.



