Increasingly, public health officials, regulatory enforcement personnel and even trade organizations are urging the global food processing industry and its supply chain partners to validate the reliability of processes used for controlling or preventing food safety failures. Salin et al.[1] reported, based on U.S. Food and Drug Administration (FDA) and U.S. Department of Agriculture (USDA) Food Safety and Inspection Service sources, that there were approximately 713 food product recalls between 2000 and 2003. The same workers report further that roughly 43% of the recall actions were initiated after the implicated food was found to contain pathogenic microorganisms. Centers for Disease Control and Prevention (CDC) data indicate that as many as 5,000 deaths (13 per day) and 325,000 hospitalizations may occur from approximately 76 million new cases of food-related illness each year in the U.S.[2] The total societal cost of foodborne illnesses in the U.S. may be as much as $152 billion per year, according to a recent report from the Produce Safety Project at Georgetown University.[3] The public health implications are enormous but so too are the implications for a company’s bottom line. Phillip Crosby, Joseph Juran and others have estimated that the cost of poor process control (poor quality) can range from 15%–40% of business cost.[4-6] The cost to eliminate a failure (defect) in the customer phase is five times greater than it is at the development or manufacturing phase. The cost for correcting food safety failures at the customer phase will be significantly higher: experience suggests, at minimum, twice as much. To paraphrase Dr. Deming,[7] do not spend your money testing and inspecting finished products. Rather, he advocated testing and inspecting the processes used for manufacturing those products. In other words, Dr. Deming was a proponent of process validation.
Why Process Validation?
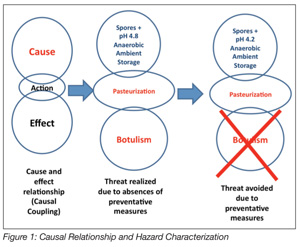 Process validation has long been an accepted way of doing business for the U.S. pharmaceutical and medical devices industries. The food processing industry has traditionally, until the advent of Hazard Analysis and Critical Control Points (HACCP), relied almost exclusively upon ex post inspection-based techniques for assessing and confirming the safety and integrity of its products. Process validation is an ex ante activity that seeks to establish the safety of a food and/or its manufacturing processes in advance of commercialization. HACCP is an ex ante food safety strategy. The outcomes, in terms of food safety, from the two approaches vastly differ. Validation is a science-based method and, when applied to food production, seeks to identify causal relationships that may have an adverse impact on the public health status of food manufactured using a specific process. That is, a set of conditions that may result in the production of an unsafe food is both examined and scrutinized to identify solutions for mitigating the identified hazard. The relationship between the hazard and its preventative measure is a causal relationship, the basis of the HACCP approach (Figure 1). While the benefits of an ex ante approach to food safety are enormous, process validation has neither been adopted by the food processing industry nor has it been demanded by regulatory agencies. Presently in the U.S., only juice, meat and seafood[8-11] are manufactured with mandatory requirements for HACCP. A series of spectacular food safety failures in the U.S. involving peanuts and peanut-derived products has had the effect of causing the FDA to compel peanut, tree nut and edible seed processors to validate the kill steps in their manufacturing processes.
Process validation has long been an accepted way of doing business for the U.S. pharmaceutical and medical devices industries. The food processing industry has traditionally, until the advent of Hazard Analysis and Critical Control Points (HACCP), relied almost exclusively upon ex post inspection-based techniques for assessing and confirming the safety and integrity of its products. Process validation is an ex ante activity that seeks to establish the safety of a food and/or its manufacturing processes in advance of commercialization. HACCP is an ex ante food safety strategy. The outcomes, in terms of food safety, from the two approaches vastly differ. Validation is a science-based method and, when applied to food production, seeks to identify causal relationships that may have an adverse impact on the public health status of food manufactured using a specific process. That is, a set of conditions that may result in the production of an unsafe food is both examined and scrutinized to identify solutions for mitigating the identified hazard. The relationship between the hazard and its preventative measure is a causal relationship, the basis of the HACCP approach (Figure 1). While the benefits of an ex ante approach to food safety are enormous, process validation has neither been adopted by the food processing industry nor has it been demanded by regulatory agencies. Presently in the U.S., only juice, meat and seafood[8-11] are manufactured with mandatory requirements for HACCP. A series of spectacular food safety failures in the U.S. involving peanuts and peanut-derived products has had the effect of causing the FDA to compel peanut, tree nut and edible seed processors to validate the kill steps in their manufacturing processes.
The FDA Food Safety Modernization Act (FSMA) has set the stage for more aggressive actions by the agency in mandating risk-based, ex ante approaches to food safety management and regulation. Of the four major provisions that comprise the basis of the FSMA, two are focused on building the agency’s capacity to both prevent and detect food safety problems, precisely the guiding principles of an ex ante approach to food safety management. A comprehensive reading of the FSMA reveals that it is written upon a framework that is anchored by food safety precepts that are process validation’s raison d’être. Specifically, Sections 101, 103 and 104 of Title 1 concern records, hazard analysis, preventive measures and performance standards. It is unambiguous to most veteran food safety practitioners that the new mantra for FDA and the food processing industry is “validation.”
U.S. processors of low-acid canned foods have long operated under the strictures of mandatory process validation as required by the Low Acid Canned Food (LACF) regulations in 21CFR108 and 113 of the Federal Food, Drug and Cosmetic Act.[12] These FDA regulations (erroneously referred to as a HACCP system) are in fact ex ante process control procedures (Good Manufacturing Practices) for mitigating the threat of botulism in canned foods. Because of their validation and implementation in 1973, the numbers of illness outbreaks associated with this class of foods have been very low for commercially processed canned foods in the U.S. From 1960 to 1971, there were, according to the CDC, 78 reported outbreaks involving 182 individuals with 42 deaths attributed to exposure to Clostridium botulinum toxin.[13] The CDC data report further that of the 42 fatalities, 26 were due to the consumption of commercially canned, low-acid foods. From 1973–1996, there were, on average, 24 foodborne botulism outbreaks per year.[14] From 1990–2000, CDC reports 160 foodborne botulism events (~10 events per year), affecting 263 people in the U.S., with an annual incidence rate of 0.1 per million. During that reporting interval, five of the 160 reported outbreaks were associated with commercially prepared and processed food, involving a total of 10 individual cases.[15] Clearly, the severe public health threat represented by improperly processed low-acid canned foods justifies the mandatory process validation requirements of the FDA’s LACF regulation.
Process Testing
It is generally understood and accepted by food safety scientists that it is impossible to achieve food safety by testing or inspecting finished products. Moreover, it is also understood that safety, quality and effectiveness must be designed and built into the product, and that each step of the manufacturing process must be controlled to maximize the probability that the finished product meets all safety, quality and design specifications. Clearly, safety can neither be tested nor inspected into the finished product. Ex post product testing and inspection regimes are generally incompatible with assuring public health expectations for safe food. The sampling plans that underpin the testing and inspection regimens almost assure the acceptance of defective materials. The possible exception is a scenario in which 100% of the products are tested and subsequently destroyed, not a practical or desired outcome for a company engaged in selling food products. This incompatibility is justification for using the more rigorous, but reliable, ex ante process validation methods.
Process validations, according to the FDA, are those collective science-based methods and procedures used for establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications for quality and food safety.[16] Though succinct, this is truly a superb and all-encompassing definition. It is replete with the requisite engineering and statistical concepts essential for process control. The demands for reliability, reproducibility and precision are its foundation. This definition is based on the requirements for established limits, specifications and capabilities, that is, validation is ultimately about control over the sources of variability, inherent or otherwise, which may impact the performance (outcomes) of a specific procedure or process. In the case of food processing, these differences in outcomes may imperil public health. Validation also demands objective data that are completely and properly analyzed.[17] Inconsistencies, vagaries and deficient information must be resolved before the validation process can be considered complete. FDA is looking for confirmation that a process is capable, also referred to as a common cause process,[7] that is, a process that can be relied upon, with a high degree of confidence, within established statistical limits, to produce the same or similar outcomes. The
benefits inuring from the use of process validation are enormous. Achieving these benefits, however, comes at considerable expense, in terms of time and money as well as demands on the business.
Validation versus Verification
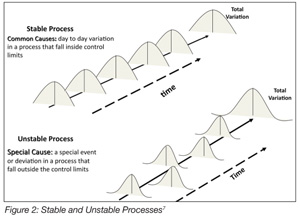 There is frequently confusion about the concepts of validation and verification. These are distinct but related activities. Verification, according to the FDA,[16] is confirmation by examination and provision of objective evidence that specified requirements have been fulfilled. Verification is the Quality Control aspect of the process. Verification presupposes that the process or procedure in question has previously been the subject of a validation study. Validation, then, is the Quality Assurance (QA) characteristic of the process. This is a critical distinction and vital to understanding the effectiveness of process validation. For example, under HACCP, there is the requirement for monitoring the process to ensure that it is operating within established critical limits. The assumption here is that the critical limits, as erected around the preventative measure, have previously been validated. That is, the HACCP team has determined that the preventative measure is capable, with a high degree of confidence, of mitigating the hazard associated with the causal relationships that define a specific CCP. Verifying the capability of a “special cause process” while assuming that it is a “common cause process” is a prescription for disaster (Figure 2). A “special cause process” is unreliable, and its outcomes are unpredictable. Dr. Deming noted two kinds of mistakes relative to common cause and special cause processes: A bad mistake is when one reacts to an outcome as if it resulted from a unique or special cause, when in actuality, it came from a common cause that will probably occur again; a catastrophic mistake, he observed, is where one reacts to an outcome as if it came from a common cause, when it actually came from a special cause.[17] A “special cause process” when used in food processing is not compatible with achieving food safety.
There is frequently confusion about the concepts of validation and verification. These are distinct but related activities. Verification, according to the FDA,[16] is confirmation by examination and provision of objective evidence that specified requirements have been fulfilled. Verification is the Quality Control aspect of the process. Verification presupposes that the process or procedure in question has previously been the subject of a validation study. Validation, then, is the Quality Assurance (QA) characteristic of the process. This is a critical distinction and vital to understanding the effectiveness of process validation. For example, under HACCP, there is the requirement for monitoring the process to ensure that it is operating within established critical limits. The assumption here is that the critical limits, as erected around the preventative measure, have previously been validated. That is, the HACCP team has determined that the preventative measure is capable, with a high degree of confidence, of mitigating the hazard associated with the causal relationships that define a specific CCP. Verifying the capability of a “special cause process” while assuming that it is a “common cause process” is a prescription for disaster (Figure 2). A “special cause process” is unreliable, and its outcomes are unpredictable. Dr. Deming noted two kinds of mistakes relative to common cause and special cause processes: A bad mistake is when one reacts to an outcome as if it resulted from a unique or special cause, when in actuality, it came from a common cause that will probably occur again; a catastrophic mistake, he observed, is where one reacts to an outcome as if it came from a common cause, when it actually came from a special cause.[17] A “special cause process” when used in food processing is not compatible with achieving food safety.
Validation Approaches
There are three strategic approaches frequently used in the validation process: prospective validation, concurrent validation and retrospective validation. Each strategy has inherent strengths and weaknesses. These are often process- and product-dependent.
Prospective validation is typically the preferred approach, especially when the process being studied has associated serious public health risk. Prospective validation is conducted prior to the distribution of either a new product or a product made under a revised manufacturing process, where the revisions may affect the product’s food safety characteristics. Novel preservation methods, aseptic processes and sterilization processes, for example, are best validated using the prospective approach. Failure modes and effects analysis (FMEA)[18,19], fault tree analysis, barrier analysis and response surface modeling methods are frequently used for prospective validation. Bio-validation techniques (e.g., inoculated packs, count reduction studies) frequently used by the food industry are excellent examples of the FMEA technique. HACCP is based largely on the fundamental principles of barrier analysis. For regulatory agencies responsible for food safety, prospective validation is the preferred method. The costs of prospective validation are likely to be high, and the time required for completing the process, protracted.
Concurrent validation methods,[20] while not suitable for a novel process, might be used for validating processing equipment that has undergone minor modification or a food system that has been reformulated. The concurrent approach is often used when there is a high level of confidence about the outcomes, and this confidence will usually be derived from data that were generated and examined under a full prospective validation of the process or product. The validation is conducted concurrent with the manufacturing process. FMEA, fault tree analysis and barrier analysis are all well suited for this. The concurrent method, if properly conducted, allows for the products that are produced under the validation protocols to be released for commercial or other applications. This is a very attractive feature of the concurrent method. It is, however, difficult to conceive of a situation in which a novel process or product can be validated using this method. The associated risk of placing an unproven product into the marketplace is simply too great.
Retrospective validation techniques are very effective and, regrettably, in common use. These methods are frequently key components of the product recall process, that is, validation of a process for a product already in distribution, based upon accumulated production, testing and control data. Root cause analysis and actionable cause analysis are techniques frequently used in retrospective validation. Typically, these methods are employed after a company receives information that has called into question the safety of a product in distribution. Executing a product recall is a stressful and hyper-pressurized event. In most instances, a product recall will cause a company to nearly suspend its normal operations. The structure and rigor demanded of the validation process will enable the recall’s successful conclusion. Retrospective validation is an ex post method that can be very useful in pinpointing a process failure. The cost of a recall involving an unsafe food typically rivals or exceeds those costs associated with either prospective or concurrent process validation.
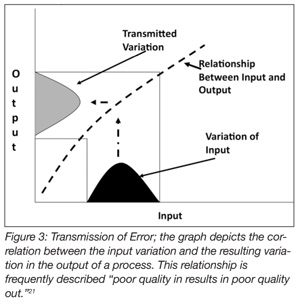 Process validation methods are also well suited for use in risk analysis (Figure 3).[21] Risk analysis is usually comprised of three conjoined activities: risk assessment, risk communication and risk management.[22] Risk assessment, among these, is science-based. Risk assessment deals ostensibly with four distinct but related activities: (1) hazard identification, the formal evaluation as to whether a specific procedure, process or product is causally linked to adverse health effects (Figure 1); (2) hazard characterization examines the relationship between the hazard and its specific outcomes; (3) exposure assessment evaluates how much of the risk factor people are likely to be exposed to and the circumstance under which the exposure is likely to occur; and (4) risk characterization integrates the data from the previous three steps and draws conclusions about whether preventative measures are required for protecting the public health. Process validation is an excellent method for vetting or confirming efficacy of the solutions that have been identified for eliminating or reducing an identified hazard to acceptable levels. In the final analysis, propositionally, food safety is, after all, risk-based.
Process validation methods are also well suited for use in risk analysis (Figure 3).[21] Risk analysis is usually comprised of three conjoined activities: risk assessment, risk communication and risk management.[22] Risk assessment, among these, is science-based. Risk assessment deals ostensibly with four distinct but related activities: (1) hazard identification, the formal evaluation as to whether a specific procedure, process or product is causally linked to adverse health effects (Figure 1); (2) hazard characterization examines the relationship between the hazard and its specific outcomes; (3) exposure assessment evaluates how much of the risk factor people are likely to be exposed to and the circumstance under which the exposure is likely to occur; and (4) risk characterization integrates the data from the previous three steps and draws conclusions about whether preventative measures are required for protecting the public health. Process validation is an excellent method for vetting or confirming efficacy of the solutions that have been identified for eliminating or reducing an identified hazard to acceptable levels. In the final analysis, propositionally, food safety is, after all, risk-based.
Steps of Process Validation
The mechanics of process validation include five steps: 1) formation of multi-functional, cross-disciplinary teams; 2) development of a master validation plan; 3) development of process validation protocols; 4) implementation of validation protocols; and 5) data analysis and reporting findings.
Step 1. The first step in the process is the assembly and training of the validation team. The validation process will typically require the skills of many. Design and execution of the validation plans will demand contributions from an assortment of disciplines. The team will ultimately be responsible for driving the validation process. For example, if a company was seeking to validate a high-intensity pulsed light process for use in decontaminating powdered milk, then one might reasonably expect that the validation team would include mechanical engineers, microbiologists, electrical engineers, food scientists, dairy scientists, physicists, sensory scientists, packaging engineers, process engineers and perhaps regulatory affairs specialists. Certainly, the activities involved in validating this process are beyond the reach of the company’s food safety and QA managers. The team members will have overall responsibility for the project, including defining the project’s scope of work, and for establishing criteria that determine success or failure. Moreover, the team will have responsibility for gaining stakeholder support for the activity.
Step 2. The importance of the master plan cannot be over-emphasized. The master plan provides a statement of the problem and identifies the strategies that will be used for defining the solution. In many respects, the plan is similar to a highly developed abstract. The master plan is the major syllogism underpinning and justifying the validation project. The plan will guide the development and execution of all subordinating activities necessary for achieving a successful outcome. It is the ultimate statement of the project’s intended scope of work. The master plan is crucial to the success of the overall validation process. Again, insofar as strategy is concerned, there are three approaches universally used for the validation process: prospective, concurrent and retrospective methods. Each strategy has upside and downside characteristics, as previously discussed. Prospective validation is completed before a process or product is manufactured using a specific process that is approved for commercial use. Hence, there is a potential associated risk for causing a delay in product launch and perhaps, consequently, losses of market share. When done correctly, however, one can move forward with absolute confidence that the product meets pre-determined specifications for both quality and food safety, thereby negating concerns (for the business) of the need for an expensive retrospective validation (product recall). The plan should also include the criteria or predetermined specifications that are to be validated. Using the hypothetical high-intensity pulsed light example, the strategy might be a prospective validation involving bio-validation (i.e., FMEA) using a surrogate organism in a series of count reduction studies. The predetermined specification, or criterion defining a successful process, might be stated as a 5-log reduction of the challenge organism/250 g powdered milk with a confidence interval of 95.999%. The master plan is typically not an overly prescriptive or detailed document. The plan may cite the relevant work of other researchers in the area and include a series of statements to justify the activity. A justification, for example, might read as follows: Salmonella-contaminated milk powder has been used in the formulation of liquid milk that is intended for use in the national school lunch program. Experts estimate on an annual basis that there are 3-4 outbreaks associated with this product, affecting between 10-35 infants and small children. The annual cost to the company, above and beyond loss of good will, is about 2.5 million dollars. The master plan may also include a budget and business case to justify the validation expense. Ultimately, the master validation plan is important for building consensus, gaining approvals and guiding the development of the requisite validation protocols.
Step 3. While the master validation plan is a strategy document, the validation protocols are tactical. They elaborate, with exquisite detail, the what, when, how and how many requirements necessary for the proper execution of the validation process. The number and complexity of the required protocols are functions of the master plan and the anticipated outcomes of the validation process. A prospective validation process, for example, involving a novel preservation method would likely require numerous protocols. In contrast, a concurrent validation scheme to confirm the efficacy of a process change may require only one or two protocols. Recall the powdered milk example in which the product was to be decontaminated using a novel, high-intensity pulsed light process. This validation effort, involving an unproven process, demands a significant number of protocols. For example, protocols would be required for the production and calibration of the bacteria used in the count reduction studies. Protocols describing the methods for inoculating the milk powder would be required. A protocol elaborating the methods for the post-treatment recovery of the bacteria would also be required. Protocols would be required for equipment setup and configuration, as well as for monitoring and measuring performance of process- critical elements. Protocols would also be required for the standardization and calibration of all devices that are used in gathering or collecting process critical data. Therefore, data loggers, thermocouples, resistance temperature detectors, pressure transducers, photo sensors and flow-rate gauges would all require calibration protocols. It is also imperative that protocols are developed for microprocessors and computer programs that drive critical aspects of the process.
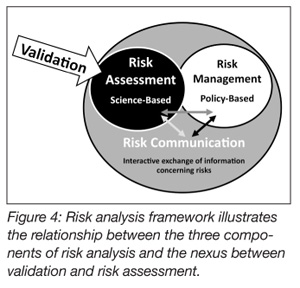 The validity and credibility of the methods employed by the protocols are also important considerations. It is difficult to validate a method or device in the context of validating a process. The methods used should be well understood and, to the extent practicable, accepted as standard methods. The methods for enumerating post-treatment bacteria, for example, may be methods that have been accepted by USDA, FDA or other authoritative bodies. If the testing methods are questioned, so too are the results obtained when using those methods, jeopardizing, potentially, the entire validation process. The same is true for non-standard measurement devices. Errors or special cause variations occurring in a process are generally not localized or isolated events. Typically, variation is transmitted across the expanse of a process. As it is often said, if the process isn’t right, the product won’t be right (Figure 4).[21]
The validity and credibility of the methods employed by the protocols are also important considerations. It is difficult to validate a method or device in the context of validating a process. The methods used should be well understood and, to the extent practicable, accepted as standard methods. The methods for enumerating post-treatment bacteria, for example, may be methods that have been accepted by USDA, FDA or other authoritative bodies. If the testing methods are questioned, so too are the results obtained when using those methods, jeopardizing, potentially, the entire validation process. The same is true for non-standard measurement devices. Errors or special cause variations occurring in a process are generally not localized or isolated events. Typically, variation is transmitted across the expanse of a process. As it is often said, if the process isn’t right, the product won’t be right (Figure 4).[21]
External review may also be critical for the overall success of the validation initiative. This is especially true when the outcomes of the validation are to withstand regulatory scrutiny. It is best to have regulatory officials involved early on in the process. It is recommended that they be offered the opportunity to critique both the master plan and protocols prior to their implementation. This is especially true for those protocols that will ultimately confirm proof or efficacy of the process. For example, if the validation were based on bio-validation, then it would benefit the validation team to seek input from regulatory officials and third-party experts insofar as the protocols relate to the desired log reduction. It would be a great transgression to present good, high-quality data to the requisite food safety authority only to have it rejected due to an apparent lack of rigor. It is also highly recommended that the team involve outside experts and representatives of the regulatory community early on in the process.
Step 4. Once the protocols are agreed upon, all team members must ensure that these protocols are executed exactly as they were intended, and this devotion to accuracy and compliance must be embraced by everyone during all steps of the validation trials. Failure to adhere to the protocols will result in great consternation for those responsible for evaluating the data.
To properly qualify the data (for step 5), one must have the utmost confidence in the manner in which the data were obtained. Data secured by methods other than those articulated in the validation protocols will confound the analysis and, ultimately, will have a negative impact on the reliability of results.
Consider the circumstance where (using the powdered milk example) the moisture level of the powdered milk, before treatment, exceeds specifications. The consequence of this out-of-tolerances condition would likely alter the survival characteristics of the challenge organism in the milk matrix and impact its kinetics of inactivation. This situation highlights the importance of raw material-defined specifications in process validation and the requirement for strict adherence to those specifications in product manufacturing. Frequently, product failures are attributed to “process failure,” when, in reality, the cause was a failed raw material specification. This misdiagnosis could have broader implications and result in significant costs to the company. The observed raw material specification non-compliance could be either a “special cause” or a “common cause” event. The assigned cause will ultimately determine the actions taken to rectify the products in inventory as well as future production. It is prudent, therefore, for food manufacturers to ensure that the raw materials used in production are within the predetermined tolerances as established by the validation protocols. This requires a raw material Quality and Food Safety program that is integral with suppliers, such that suppliers’ shipping docks become the food manufacturer’s receiving dock.
In another example, consider the implication of an equipment setup where the pulser for the high-intensity light system was set to operate at a repetition rate faster than specified in the protocol, resulting in an increase in total radiant energy (J/m²) delivered at the point of inactivation. The corresponding, but unexpected, increased lethality would likely cause great confusion for those charged with analyzing the data. In other words, confidence in the data is absolutely dependent on the confidence that the protocols were properly executed.
Another important consideration related to implementation of the protocols has to do with reproducibility. That is, the validation trials must be repeated enough times to allow a determination that the data generated during the trials are reproducible. Reproducibility is among the first principles underpinning the scientific method. One or two sets of experiments, for sure, will not answer this fundamental but important question. The validation team and its leader must establish the minimum number of trials essential for the unambiguous demonstration that the data are both robust and reproducible with a high degree of confidence.
Step 5. The penultimate and perhaps the most daunting aspects of the validation process are data analyses and issuance of a proclamation of success or failure. Every particle of data must be collated and examined, all mysteries solved and anomalies explained. This process too should be a team activity. Each member should bring their respective skills to scrutinizing and teasing out the subtleties and nuances of the data that may affect the manner in which they are interpreted. Computer analyses of certain data are routine and a great aid in moving the process forward. These data must also be evaluated by the validation team, however, to ensure, to the extent practical, that they are reasonable and correct. Computer programs have been found to contain errors. Mentioned previously was the importance of utilizing proven methods and technologies in all phases of the validation process. This axiom also applies to computer programs.
The ultimate step of the process involves the reporting of relevant findings. Obviously, reporting requirements are project specific. The stakeholders may be a company’s senior management, marketing managers, public health authorities or regulatory and enforcement officials. The context in which the validation was initiated will determine how it is reported. If the work were done on a proprietary basis for use by a single company, then the team would report, logically, to its management team. By contrast, if the report were intended for a broader audience, say perhaps the regulatory community, then the team must assure that the reporting mechanism is compatible with the agency’s requirements and expectations. For example, if the validation were done as part of the requirements for a mandatory HACCP program, then the regulation governing that program would dictate how the results are reported. Likewise, if the validation were performed to support or underpin a LACF Petition, then FDA’s regulations, including the preparation of a 2541a, would determine how the data are reported. Generally speaking, it is the quality of the data as opposed to the volume of the data that is important. The report should answer the initial questions that were posed in the master validation plan. Likewise, the report should also qualify the solution(s) that result from the study. For example, if the proposed solution was a 5-log reduction of surrogate organisms, then the report would need to confirm that the surrogate was the right organism for the project and that the correlation between its inactivation and that of the pathogen is positive and will allow, therefore, a reliable estimation of risk to ensuring public health.
Where Do We Go from Here?
Ex post food safety procedures, based solely on testing and inspecting finished products, were long ago shown to be ineffective. The safety of foods intended for the U.S. spaceflight program were deemed high risk, not suitable for this activity, because their food safety status was based on ex post finished product testing. It was concluded, at that time, that to achieve the confidence necessary to allow such foods in space would require 100% inspection (destructive testing) of the products. Obviously, this was not a tenable solution. Subsequently, the National Aeronautics and Space Administration and its supply chain partners in the food processing industry recognized the benefits of an ex ante HACCP approach to food safety. In other words, it was concluded that the benefits, in terms of confirming food safety, were derived from testing and inspecting the processes that were used in food manufacturing rather than testing the finished product. Drs. Deming and Bauman, quite independent of each other, advocated process validation for food processing operations. The spectacular food safety failures of the last decade suggest that the food industry has not come to grips with this reality. The QA offices of many companies are filled with HACCP plans that are built on faulty assumptions about the capability of the plan’s preventative measures, allowing, therefore, the fruition of a causal situation that could result in illness or injury to those exposed to products manufactured with the process. The preventative measures, in many instances, have not been properly validated. Food companies are still relying on ex post testing of finished products as a means of conferring food safety. As illustrated above, food safety cannot be tested or inspected into finished product; achieving food safety is dependent on process control capabilities.
It is clear that as we advance and embrace new technologies, new methods and new systems for supporting the mass production of human food, we must provide new thinking and new methods for ensuring its safety. Obviously, the food industry and regulatory agencies would much prefer the outcomes of a prospectively validated process than the use of retrospective methods to retrieve potentially harmful foods from the distribution channels. Food safety is a risk-based activity, defined as the biological, chemical or physical status of a food that will allow its consumption without incurring excessive risk of injury, morbidity or mortality. If we accept this premise, then we must also acknowledge that achieving food safety has to do with assessing risk. FDA’s FSMA is constructed around this premise. The legislation is reflective of the fact that FDA understands, fundamentally, that mitigating food safety failures is predicated on identifying failure modes, understanding the likelihood of an event’s occurrence and providing prevention. To be effective, however, the underlying assumptions for failure and prevention must be properly validated.
Process validation is an effective risk assessment strategy. When these methods were applied to food production, as in the LACF regulations, a precipitous and sustained improvement in the food industry’s ability to manage the serious risk of botulism associated with this class of foods was the result. Process validation is ultimately about the data. Having good data is like having a crystal ball. It has been said that data can enable a view to past performance, confirm present operating conditions and allow a confident projection about future performance. Achieving food safety cannot be legislated nor can it be assured using ex post testing and inspection methods. It is reasonable, however, with a high degree of confidence, to expect safe food as an outcome when properly designed and carefully controlled systems of manufacturing are used in its production.
Larry Keener is president and chief executive officer of International Product Safety Consultants, Inc. (IPSC). He is an inaugural member of Food Safety Magazine’s editorial advisory board.
Jerry Roberts is an accomplished executive leader within the consumer packaged goods industry. He is a long time member of the Food and Nutritional Science Advisory Board at Tuskegee University.
References
1. Salin, V., S. Darmasena, A. Wong and P. Luo. 2005. Food product recalls in the USA, 2000–2003: Research update presented at the Annual Meeting of the Food Distribution Research Society.
2. www.cdc.gov/ncidod/eid/vol5no5/mead.htm.
3. www.producesafetyproject.org/admin/assets/files/Health-Related-Foodborne-Illness-Costs-Report.pdf-1.pdf.
4. Crosby, P. 1979. Quality is free: The art of making quality certain. New York: McGraw-Hill.
5. Crosby, P. 1985. Quality improvement through defect prevention. Phillip Crosby and Associates.
6. elsmar.com/wiki/index.php/Juran.
7. Deming, W. E. 1993. The new economics for industry, government, education. Cambridge, MA: MIT Press.
8. Keener, L. 1999. Is HACCP enough for ensuring food safety? Food Testing and Analysis, 5.
9. Keener, L. 2001. Buying into HACCP. Meat Processing Magazine September.
10. Keener, L. 2001. HACCP: A view to the bottom line. Food Testing and Analysis.
11. U.S. Code of Federal Regulations. 21CFR 123 - FDA Seafood HACCP Regulations.
12. U.S. Code of Federal Regulations. 21CFR 113 - FDA Low Acid Canned Foods Regulations.
13. www.cdc.gov/ncidod/dbmd/diseaseinfo/files/botulism.pdf.
14. Shapiro, R. L., C. Hatheway and D. L. Swerdlow. 1998. Botulism in the United States: A clinical and epidemiologic review. Ann Intern Med 129(3):221-8.
15. www.cdc.gov/ncidod/EID/vol10no9/03-0745.htm.
16. www.complianceassociates.ca/pdf/Guide_-_Process_Validation.pdf.
17. Shewhart, W. A. 1986. Statistical methods from the viewpoint of quality control. New York: Dover Publications.
18. McDermott, R.E. et al. 1996. The basics of FMEA. Portland, OR: Productivity Press.
19. Dailey, K.W. 2004. The FMEA pocket handbook. DW Publishing Company.
20. Taylor, W.A. 1998. Methods and tools for process validation: Global Harmonization Task Force Study Group #3. Taylor Enterprises.
21. Hojo, T. 2004. Quality management systems: Process validation guidance, Global Harmonization Task Force, Study Group #3, Edition 2.
22. Cole, M., D. Hoover, C. Stewart and L. Keener. 2011. New tools for microbiological risk assessment, risk management, and process validation methodology. In: H. Q. Zhang et al. (eds.) Nonthermal processing technologies for food. Wiley Press.
Ex Ante or Ex Post Food Safety Strategies: Process Validation versus Inspection and Testing




