
Even though the emphasis in the food industry during the past several years has been on microbial safety and food defense, it is also important to note that significant issues arising from chemical contaminants and allergens in foods continue to require industry’s serious attention. There have been a number of product recalls, public health alerts and trade issues on a worldwide basis related to chemical toxins, both man-made and naturally occurring, in the past few years. These range from alcohol to perchlorate, to Sudan Red dye to acrylamide.
As stakeholders in food safety, we want to take a look at these potentially harmful chemical contaminants to see what can be done to prevent or mitigate any adverse affects such compounds do or might have on public health, the food business and global trade. This means that we need to conduct appropriate food safety research in order to establish scientifically sound food safety public policy. By “appropriate” food safety research we mean supporting ethical research, promoting significant reproducible research findings to the public so we can share the benefits of the research supported by taxpayer monies, and preventing abusive use of research results. We want to establish regulations to achieve all of the above, and all of these regulations ought to be based on science so that all food safety decisions are, in fact, science based.
Regulating for food safety improvement leads to many benefits for consumers, including improved diets with reduced risks and opportunities to enjoy the benefits of health research. On the business side, industry benefits from the minimization of unfair competition and is provided with opportunities to develop enhanced products. Of course, realizing these benefits requires us to develop very clear definitions of what is healthful, what is harmful and at what level, scientifically speaking, we can say with some confidence that a compound is either “positive for” or “adverse to” one’s health. Only with clear, science-based definitions will food safety regulations truly be relevant.
From the food research laboratory’s perspective, this requires adequate, relevant analyses that support the definitions needed for good policy making. In other words, we must look critically at the combined issues and analytical aspects of detection monitoring and controlling chemical contaminants in food and ask: How do we analyze these compounds? What is the best way? And what should we be shooting for in terms of detecting relevant lower detection limits?
Measuring Absolute, Chasing Zero
As food chemists, we work with two terms that will affect the ultimate answers to these questions. Though used rarely, the term “absolute” is often used in reference to the verified purity of chemical reagents and solutions used in the analytical laboratory and/or to denote the direct analysis of some established high purity food ingredients to verify these materials as pure, such as alcohol, sucrose, salt and lactose. Generally speaking, however, absolute purity of a food is usually determined by a lack of contaminants in that food. The U.S. Pure Food and Drug Act mandates that all foods should be pure although, chemically speaking, no single food in existence is absolutely pure. The law is necessarily interpreted as “Thou shalt not adulterate or contaminate your food.”
 In reality, then, we do the opposite of measuring the absolute: We actually measure the undesirable impurities that are in the food and determine the food to be pure when such impurities are not present, or if present, below a regulatory based quantity. Which brings us to the other important term relevant to this discussion: “zero.” How low should our detection limits go to determine whether at those impurities are, in fact, present? With each decade, we keep approaching a “smaller zero” and zero keeps pushing back. Zero is actually a small number that has a very big impact, as shown in Figure 1. Take a look at what happens when we start with a 100% pure material where we have just a single compound to analyze. As we progress down the scale of possible lower levels of detection—parts per ten (ppten), parts per hundred (pph), parts per thousand (ppth), through parts per quadrillion (ppq)—the number of compounds we must potentially look at grows exponentially. When we are measuring the material at the parts per trillion (ppt) level, we may now have 1015 to 1018 possible compounds that could be identified and quantitated, most of them present at very low levels—and most of these likely insignificant relative to how we define purity.
In reality, then, we do the opposite of measuring the absolute: We actually measure the undesirable impurities that are in the food and determine the food to be pure when such impurities are not present, or if present, below a regulatory based quantity. Which brings us to the other important term relevant to this discussion: “zero.” How low should our detection limits go to determine whether at those impurities are, in fact, present? With each decade, we keep approaching a “smaller zero” and zero keeps pushing back. Zero is actually a small number that has a very big impact, as shown in Figure 1. Take a look at what happens when we start with a 100% pure material where we have just a single compound to analyze. As we progress down the scale of possible lower levels of detection—parts per ten (ppten), parts per hundred (pph), parts per thousand (ppth), through parts per quadrillion (ppq)—the number of compounds we must potentially look at grows exponentially. When we are measuring the material at the parts per trillion (ppt) level, we may now have 1015 to 1018 possible compounds that could be identified and quantitated, most of them present at very low levels—and most of these likely insignificant relative to how we define purity.
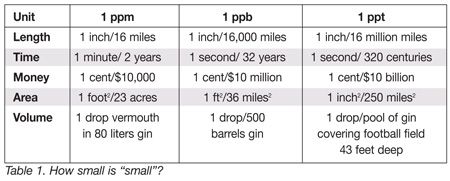 Now, the question is: How low in concentration do we go? To gain a bit of perspective on what we are measuring, consider Table 1, which illustrates by use of standard measurement units such as length and volume just how small “small” is, comparatively speaking, when considering ppm, ppb and ppt, common units of measurement used to determine impurities (or lack thereof) in foods. In terms of length, if 1 inch represents 1 ppm, then the distance we are considering is 16 miles. When we look at 1 ppb, we are measuring 1 inch in 16,000 miles (approx distance around the world at the latitude of Minneapolis/St. Paul, Minnesota). It isn’t difficult to see that when we drop down to parts per trillion, it’s the comparison between an inch and 16 million miles (or equivalent to shuffling your feet 6 inches versus taking a giant leap from here to the sun). In terms of time, it is the difference between 1 minute in 2 years versus 1 second in 320 centuries when comparing ppm and ppt. Finally, if we want to fix ourselves a “very dry” martini (ppt level), we need only to add one drop of vermouth to a pool of gin that covers the area of a football field and is 43 feet deep.
Now, the question is: How low in concentration do we go? To gain a bit of perspective on what we are measuring, consider Table 1, which illustrates by use of standard measurement units such as length and volume just how small “small” is, comparatively speaking, when considering ppm, ppb and ppt, common units of measurement used to determine impurities (or lack thereof) in foods. In terms of length, if 1 inch represents 1 ppm, then the distance we are considering is 16 miles. When we look at 1 ppb, we are measuring 1 inch in 16,000 miles (approx distance around the world at the latitude of Minneapolis/St. Paul, Minnesota). It isn’t difficult to see that when we drop down to parts per trillion, it’s the comparison between an inch and 16 million miles (or equivalent to shuffling your feet 6 inches versus taking a giant leap from here to the sun). In terms of time, it is the difference between 1 minute in 2 years versus 1 second in 320 centuries when comparing ppm and ppt. Finally, if we want to fix ourselves a “very dry” martini (ppt level), we need only to add one drop of vermouth to a pool of gin that covers the area of a football field and is 43 feet deep.
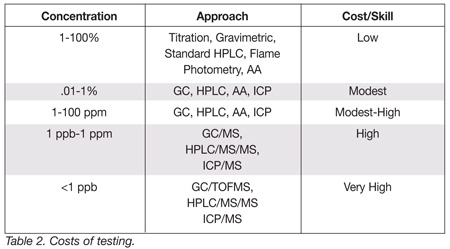 It is obvious, then, that these low level assays require higher levels of analytical selectivity and sensitivity because we are looking for ever decreasing quantities amongst so many compounds that show up at very low levels. Again, as shown in Figure 1, at low levels of detection the analyst isn’t just looking at one or two compounds anymore, even though that may superficially appear to be the case with selective detection systems. So, how much analytical selectivity and sensitivity is enough, and can we afford it? If we look at some of the traditional analytical methods used to determine percentages of target compounds in food samples, such as titration, gravinimetry, standard high performance liquid chromatography (HPLC), flame photometry and atomic absorption spectrophotometry (AA), the costs in terms of the instrumentation and analyst’s skill level tend to be relatively low (Table 2). When we drop below a percent, the costs increase modestly. If we need to detect to the lower parts per million level, even though we are able to use the same common instrumentation, we experience a modest to high cost since the analytical skill level required is increased. Drop to a ppb level and the costs begin to increase more substantially because we now require more sensitive hybrid equipment, such as gas chromatography/mass spectrometry (GC/MS), HPLC/MS, and inductively coupled plasma spectrometry-mass spectrometry (ICP/MS), instruments which can handle the exponentially increasing number of compounds that will be detected at ever lower concentration levels. And finally, if we get much below a ppb, we will need to use the newest state-of-the-art equipment: GC with time of flight MS (GC/TOFMS), HPLC/MS/MS and ICP/MS. For these analyses, the cost is very high, both in terms of the capital equipment investment and from an analytical skill perspective.
It is obvious, then, that these low level assays require higher levels of analytical selectivity and sensitivity because we are looking for ever decreasing quantities amongst so many compounds that show up at very low levels. Again, as shown in Figure 1, at low levels of detection the analyst isn’t just looking at one or two compounds anymore, even though that may superficially appear to be the case with selective detection systems. So, how much analytical selectivity and sensitivity is enough, and can we afford it? If we look at some of the traditional analytical methods used to determine percentages of target compounds in food samples, such as titration, gravinimetry, standard high performance liquid chromatography (HPLC), flame photometry and atomic absorption spectrophotometry (AA), the costs in terms of the instrumentation and analyst’s skill level tend to be relatively low (Table 2). When we drop below a percent, the costs increase modestly. If we need to detect to the lower parts per million level, even though we are able to use the same common instrumentation, we experience a modest to high cost since the analytical skill level required is increased. Drop to a ppb level and the costs begin to increase more substantially because we now require more sensitive hybrid equipment, such as gas chromatography/mass spectrometry (GC/MS), HPLC/MS, and inductively coupled plasma spectrometry-mass spectrometry (ICP/MS), instruments which can handle the exponentially increasing number of compounds that will be detected at ever lower concentration levels. And finally, if we get much below a ppb, we will need to use the newest state-of-the-art equipment: GC with time of flight MS (GC/TOFMS), HPLC/MS/MS and ICP/MS. For these analyses, the cost is very high, both in terms of the capital equipment investment and from an analytical skill perspective.
Therefore, we need to consider how and when we chase this zero level. When a contaminant is found in food and deemed to be unacceptable, we typically set zero at whatever constitutes the current limit of detection (LOD) at that point in time. When an instrumentation manufacturer introduces a new analytical instrument this often results in a significant drop in the LOD, and with the lower detection we inevitably detect more and more compounds as illustrated earlier. Therefore, the contaminant may be found to be present again at lower levels, and new contaminants may be detected for the first time. This is our challenge: As we detect more compounds and report on the findings, society as a whole, because they’ve heard bad things about some of these compounds feels obligated to chase that receding zero despite the fact that the risk level was considered and regulations set based on data using the previous, higher detection limit. Can science draw a line at what we should be looking at in terms of risk and how we handle setting appropriate limits for these compounds? Should we be chasing zero in all cases?
Cases in Approaching Zero
There are many cases in recent years in which food chemists and researchers have engaged in approaching zero, including packaging residues such as bisphenol A (BPA), isopropylthioxanthone (ITX) and butadiene; processing residues, such as chloropropanols from acid-hydrolyzed vegetable proteins; heavy metals in foods, such as mercury in fish, cadmium in vegetables and lead in chocolate and water; and mold toxins, such as aflatoxin B1, fumonisins B1, and deoxynivalenol (DON) in cereal grains, nuts and oil seeds. Again, we can look at these chemical compounds critically and ask: How do we best analyze such compounds, and what detection levels should we be shooting for in order to better define the risk to public health posed by these chemical contaminants and thus develop appropriate public health policies? Let’s consider some well-known cases of approaching zero in chemical contaminant analysis that illustrate the challenges we face in answering these questions:
Chloramphenicol in Honey. In 2002, the U.S., Canada and Europe were alerted the fact that imported honey and honey products from a leading global supplier of these products were found to contain trace amounts of chloramphenicol, a potent bacteriostatic antibiotic whose use is typically limited to treating typhoid fever and other antibiotic-resistant infections in humans, and in veterinary medicine it is often used to treat cattle suffering from Salmonella infections. Bee hives had been treated with this antibiotic and others in 1997, when an outbreak of a bacterial disease threatened to force the destruction of all hives to stop the spread of the outbreak—and along with it, the market-leading honey industry.
However, since chloramphenicol had been declared carcinogenic by the U.S. and others by 2002, it was deemed an unacceptable substance for use in production of food products. At the time, sampling of honey from the leading global supplier showed trace amounts of 0.3 to 34 ppb chloramphenicol in most of the samples, using a test method with a typical detection limit of 0.3 to 0.5 ppb.
When the world market began to be flooded with low-priced, below-cost honey products, the U.S. took note and applied stiff tariffs on to these honey imports. When exporting companies tried to avoid paying these tariffs by distributing honey products through other countries, testing was instituted to identify and trace which products contained chloramphenicol in order to remove them from worldwide distribution.
According to food safety regulatory authorities, at this point in time we are not able to set an acceptable level for chloramphenicol for public health purposes and as such, the compound is banned from use in any food products. Accordingly, the majority of food manufacturers that make products using honey or royal jelly as a component of finished products are typically analyzing samples with methodology have an LOD for honey down to 0.3 to 0.5 ppb.
At that time, manufacturers were (and still are) able to use an automated analyzer with a chloramphenicol-specific test kit that is relatively inexpensive to use in which the sample is dissolved in water, a reagent tablet is added followed by incubation, centrifuge and resuspension. The analyst is able to read the results, with a LOD of 0.3 to 0.5 ppb, on the instrument. Later, Canadian scientists improved that LOD to 0.05 ppb, or 50 ppt, using LC/MS/MS. The LC/MS/MS technique involves diluting the honey 50:50 with water, then extracting into EtOAC (2X), followed by centrifugation, evaporation, extraction into hexane, additional centrifugation, then clean up on conditioned SPE cartridges, followed by evaporation and taking up in 0.1% formic acid. The sample is then run on the LC/MS/MS instrument, resulting in very specific, highly sensitive test results, with a consequent drop to 50 ppt LOD but with a substantial increase in cost and complexity.
So, what happens in a case such as this, of chasing zero? It causes upset to business, regulators, and to some extent, probably erodes consumer confidence because we’ve discovered a potential problem but do not know to what detection level chloramphenicol should be tested and thus whether the investment in methodology we’re using is warranted. Consumers might view this lack of knowledge as a red light to consuming honey. In this case, chasing zero can also affect the extent of testing as well. With industry and government using kit test methodology at the height of the imported honey incident, extensive sampling and testing of honey and hive-derived products was being carried out to ensure that product contained no detectable chloramphenicol. With the more sensitive and sophisticated LC/MS/MS instruments being utilized by a small number of laboratories that can afford to purchase the technology and hire analysts with higher skill sets to operate them and interpret results, the volume of testing for chloramphenicol logically decreases. Is the food supply safer today, then as a result of the greater method sensitivity? The answer is as yet unknown.
Acrylamide in Foods. In chasing zero for this industrial chemical, we’ve discovered important information about how acrylamide forms and begun intensive research to identify ways to reduce its incidence in foods. Acrylamide, typically associated with industrial processes and production of such materials as plastics, grout and sealants, is known to be a neurotoxin in humans and carcinogenic to animals at ppm levels. In 2000, it was found in laboratory rat chow, but acrylamide wasn’t traced to foods until 2002 when a group of Swedish researchers announced results from a study that showed for the first time the unintentional formation of measureable levels of the chemical contaminant during the frying or baking of potatoes and cereal products. This caused public health concerns on a global basis, although the true health impact of acrylamide levels in foods was, and still is, in essence, unknown.
According to a subsequent report by a United Nations Food and Agriculture Organization (FAO) and the World Health Organization (WHO) Expert Committee, acrylamide is formed when certain foods, particularly plant-based foods that are rich in carbohydrates and low in protein, are cooked at high temperatures such as in frying, roasting or baking, generally at temperatures above 120C. The major foods contributing to acrylamide exposure in countries for which data were available are potato chips, coffee and cereal-based products, such as pastries, breads and rolls. Since constituent makeup, cooking temperature, and time affects the amount of acrylamide formed, (quantities often varying widely in the same foods or food categories), scientific experts have stated that it is not possible to issue recommendations regarding food comsumption guidelines with regard to acrylamide content. The experts did recommend that additional studies were necessary in order to fully evaluate the toxicity and health impact of acrylamide exposure through foods.
Relevant analytical techniques to detect and quantitate acrylamide in foods are available, such as GC and GC/MS methods following derivitization, and LC/MS/MS, the latter appearing to be the current instrument of choice due to the straightforward analytical procedure, as follows:
• Add internal standard (D3 Acrylamide) to sample
• Dissolve/extract sample by shaking
• Centrifuge at 9000 rpm, draw off aqueous phase
• Centrifuge at 7000 rpm through membrane filter
• Clean up on two SPE cartridges
The sample extract is loaded into the LC/MS-MS instrument and undergoes separation using an acetic acid/ methanol/water mobile phase with electrospray introduction to the MS. The analyst then monitors the transition from 72fi55 for acrylamide, as well as the transition from 75fi58 for the D3 acrylamide, and then uses the two transitions compared to calibration standards to quantitate. With this method, an LOD of approximately 10 ppb can be achieved.
So, we have the testing methods to detect and quantify acrylamide at the parts per billion level, but the question remains, does the acrylamide impurity being formed during cooking, frying, and baking pose a toxicity risk to humans consuming it in food? We know that acrylamide been in the diet for a long time, perhaps increasing in quantity consumed about 50 years ago with the mass introduction of French fries and potato chips to the U.S. population. Consumption of this chemical substance increased in the 1950s, as fast food restaurants and convenience snack foods rose in popularity. Still, no one knows quite what level of intake of foods containing detectable acrylamide results in a risk to human health.
Certainly, the quest to quantify this risk has spurred extensive product and process testing, including major international efforts to look at formation mechanisms and learn how to reduce its formation in foodstuffs, and brought a regulatory focus on developing interventions. The positive aspect is that scientists have good analytical techniques at their disposal to approach the elusive zero of acrylamide—methods used to show that acrylamide is primarily formed when reducing sugars react with asparagine in the presence of heat. It appears, generically speaking, that production of a 100 ppb of acrylamide, occurs when 100 ppm of asparagine are heated in the presence of 10 ppth of glucose—with the production being limited by limiting either of the two starting components or reducing the heat.
Thus, while the toxicity testing results are still being obtained, some reduction in consumption by reduction of acrylamide formation during food production has already been achieved due to the knowledge we’ve gained utilizing good analytical tools. In other words, our levels of consumption of acrylamide have gone down because the production/cooking processes of some fried or baked foods have been adjusted according to new information about how the substance forms in foods. Debate continues, of course, as other studies show that sources other than foods may be equally great contributors in terms of acrylamide intake, but from an analytical perspective, sampling and testing will continue as scientists pursue fuller knowledge of acrylamide formation and content of foods.
Sudan Red/Para Red. The family of azo dyes, including red dyes Sudan I, II, III and IV, Sudan-Red B, 4-(Dimethylamino)azo benzene (DMAAB) and Para Red—is most commonly used as coloring agents in the textiles and leather industries. These dyes are deemed carcinogenic and long ago were disallowed for use in any food by virtually every nation in the world. According to a white paper by the American Spice Trade Association, “Sudan Red and Related Dyes,” in 2003, ground capsicums (hot chilies) produced in India were found to contain Sudan I at levels up to 4,000 ppm. Unfortun-ately, the illegal dye was being intentionally added to chilies, paprika and other red-toned spices to enhance the food’s visual, surface appearance. New sampling and testing requirements were mandated for hot chili products. Although the exporters in question had their national certificates of registration revoked by the Indian authorities, other cases of purposeful addition of Sudan Red (and later, Para Red) dyes to food products occurred shortly thereafter. In 2004, for example, the illegal dye was detected in unrefined palm oils, and in 2005, authorities discovered Sudan Red I in U.K.-manufactured Worcester sauce, a discovery which forced the largest recall in history of the bottled sauce and food products containing the sauce as a flavoring ingredient. Throughout 2005, Sudan I-containing ingredients and foods were recalled in several nations, including the Canada, South Africa and China, and in the U.K., two food products were recalled after the detection of Para Red dye in tested samples.
While the European Union Commission has called for a limit of detection of 0.5-1.0 ppm using HPLC for Sudan I and similar chemical dyes, U.S. regulators have not yet issued recommendations on LOD for these dyes. Even so, these dyes can be analyzed in food samples using a single Soxhlet extraction, followed by size exclusion cleanup, HPLC or LC with ultraviolet-visible (UV-Vis), photo diode array (PDA). MS, or MS/MS (using electrospray introduction) detection , to obtain a LOD of 10 ppb for Sudan dyes and DMAAB and 100 ppb for Para Red.
This is clearly a case in which the lowest possible level of detection is in order. Stopping purposeful adulteration of food—perpetuated solely for illicit economic gain and without regard for placing people at risk—makes is critically important that we approach zero. The lower the detection limit we can achieve on this type of test, the better off we are, in light of the fact that quick responsible actions can be taken to stop the adulteration.
Should We Chase Zero?
What we can learn from these chemical contaminant case studies about chasing zero? First, we can see that some undesirable substances are unintentional (and in some cases, unavoidable) additions to the food as a result of processing and manufacturing, some chemical contaminants are naturally occurring in the food supply, and some are intentionally added into foods but are now unavoidable at levels close to zero. Some are carcinogens or metals that are of concern to health, and some chemicals pose high exposure while others do not. Also, we see that regulatory mandates differ in some cases from what the science tells us in terms of appropriate detection limits.
As we approach zero, we generate anxiety because we keep finding more and more compounds but since these additional compounds are detected at lower and lower levels, how much of a food safety hazard do they pose to human health? Dose makes the poison but it can also make the medicine. As we get to these extremely low levels, we also have a lot less risk. So how much should we be concerned about? And how do we, as a society and in the testing laboratory, allocate limited resources to pursue all of the possible issues raised when we approach zero? We will need to draw some lines and apply our best technology. But which action is more responsible: Should we use test kits that have a broad application but a higher LOD? We can do a lot of testing at the elevator door with this strategy but when the trucks arrive at the receiving dock are we missing adulterants which we might have picked up using a method with a lower limit of detection. Or, do we use high sensitivity, low LOD tests and do less testing because capital and skill costs rise more than the budget will allow, but miss potential adulterants that we might have picked up with broader sampling and testing using lower cost methods? Which protects the public health the best?
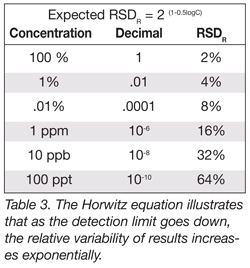 These questions are important to consider and one way that analytical food laboratories can contribute to the answers is to test methods through collaborative studies between labs to ensure that good methods are developed and implemented, and that poor methods are ferreted out. From an analytical perspective, achieving agreement between labs on contaminants present at very low levels can be a challenging process. Former U.S. Food and Drug Administration scientist Dr. Bill Horwitz devised an equation that illustrates this (Table 3). The Horwitz Equation looks at a method’s standard deviation in relative terms; i.e, the variability of the results between laboratories divided by the quantity of analyte present. Note that as the detection limit goes down, or more specifically, the quantity measured goes down, the variability increases exponentially. For example, if we have a pure material (100%) and we send it out to several laboratories for analysis, we expect the relative standard deviation (RSD) to be +/-2%, or the results reported to be 100+/-2%.
These questions are important to consider and one way that analytical food laboratories can contribute to the answers is to test methods through collaborative studies between labs to ensure that good methods are developed and implemented, and that poor methods are ferreted out. From an analytical perspective, achieving agreement between labs on contaminants present at very low levels can be a challenging process. Former U.S. Food and Drug Administration scientist Dr. Bill Horwitz devised an equation that illustrates this (Table 3). The Horwitz Equation looks at a method’s standard deviation in relative terms; i.e, the variability of the results between laboratories divided by the quantity of analyte present. Note that as the detection limit goes down, or more specifically, the quantity measured goes down, the variability increases exponentially. For example, if we have a pure material (100%) and we send it out to several laboratories for analysis, we expect the relative standard deviation (RSD) to be +/-2%, or the results reported to be 100+/-2%.
As shown in Table 3, if we are measuring a concentration of a part per million, the expected RSD increases to +/-16%, and the results reported would be 1.00 +/- 0.16 ppm. If we are measuring a concentration of a part per billion, we increase to 32% RSD. At this point, if we are getting reports from numerous laboratories, we start getting a few results that will be reported a non detected. By the time we get down to 100 ppt concentration, our estimated relative standard deviation will be 64% and approximately 15% of the results will actually be recorded as zeros or nondetected.
It is easy to see, then, that we do face a real challenge in enforcing regulations because when we get down to these low levels there is significant variability in analytical results. That variability is anathema to achieving solid science-based data from which we can set realistic limits, ensure that analytical methods conform to those limits, and contribute to good food safety policy making and good decisions about where to allocate resources to best protect human health and well-being.
Jonathan W. DeVries, Ph.D., is Senior Principal Scientist at General Mills Inc. and the Technical Manager of the Medallion Laboratories subsidiary of the company, which provides analytical services to the food industry. During his career DeVries has served on the Official Methods Board of Association of Official Analytical Chemists International (AOAC); on the Board of Directors of AOAC; and as a chair of the Committee of Food Industry Analytical Chemicals of the National Food Processors Association.
DeVries is currently chair of the Technical Advisory Committee of the National Center for Food Safety and Technology, and is chair of the Carbohydrates Technical Committee of the International Life Sciences Institute (ILSI) North America. He is a fellow of AOAC International and member of the American Association of Cereal Chemists, American Society for Testing and Materials, and the American Chemical Society. He is a founding member of Food Safety Magazine’s Editorial Advisory Board.


